
Machine Learning
Skin care
peer-reviewed
Screening Antioxidant Ingredients Using Quantum Mechanics and Machine Learning
HAIDONG LIU*, ALEX K. CHEW, RYNE C. JOHNSTON, JEFFREY M. SANDERS, ANDREA BROWNING, THOMAS HUGHES, DAVID GIESEN, MATHEW D. HALLS
*Corresponding author
Schrödinger Inc, New York, NY, United States
ABSTRACT:Antioxidants are integral ingredients for personal care products, as they inhibit the formation of free radicals that often lead to cell damage and chronic diseases. Though there is increasing consumer awareness and demand for more sustainable natural antioxidants in cosmetics products, the high-throughput screening for new antioxidant candidates is challenging and expensive, often performed experimentally using trial-and-error approaches; alternative computational approaches that could help screen antioxidant candidates are necessary. In this work, we demonstrated an efficient computational approach that uses quantum mechanical (QM) calculations and graph-based machine learning tools to predict the hydrogen bond dissociation energy, which dictates the propensity of antioxidants to neutralize free radicals. We then constructed a library of antioxidant molecules with the calculated hydrogen dissociation energies based on QM. The machine learning model was then trained and validated on the dataset to predict the hydrogen dissociation energies. This workflow enables the rapid screening of large molecule datasets to identify promising antioxidant candidates at a reduced cost by providing molecular insight and removing the need for large scale experiments.
??????????????????
“
“A study in healthy women providing probiotic yogurt for four weeks showed an improvement in emotional responses as measured by brain scans”
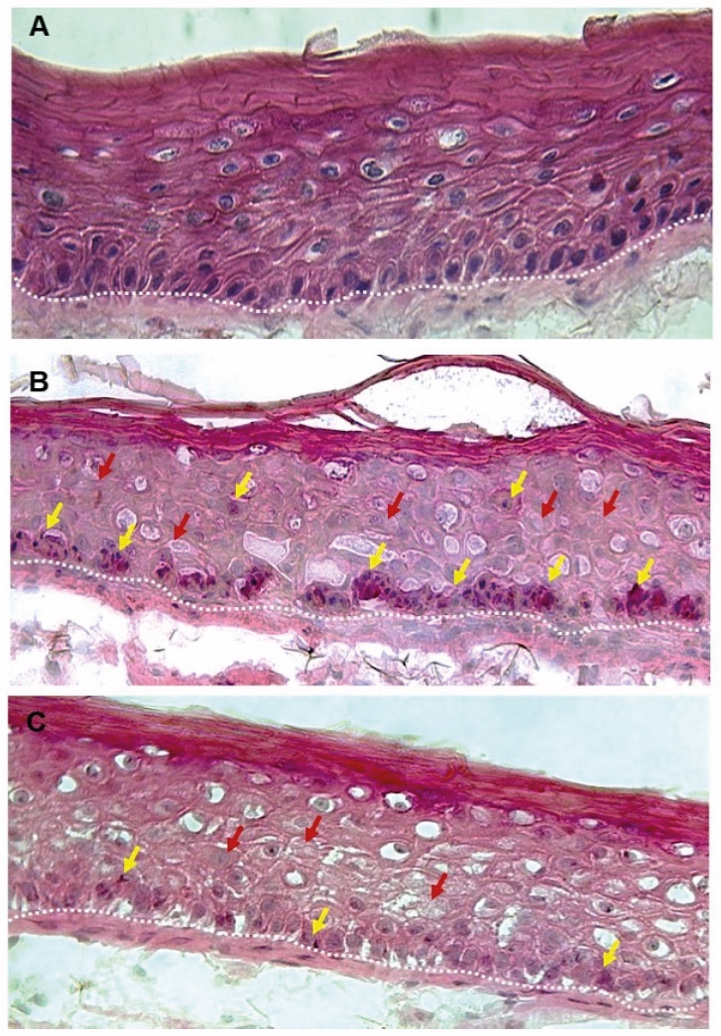
Figure 1. Skin Section with Microbiome. Most microorganisms live in the superficial layers of the stratum corneum and in the upper parts of the hair follicles. Some reside in the deeper areas of the hair follicles and are beyond the reach of ordinary disinfection procedures. There bacteria are a reservoir for recolonization after the surface bacteria are removed.
Materials and methods
Studies of major depressive disorder have been correlated with reduced Lactobacillus and Bifidobacteria and symptom severity has been correlated to changes in Firmicutes, Actinobacteria, and Bacteriodes. Gut microbiota that contain more butyrate producers have been correlated with improved quality of life (1).
A study in healthy women providing probiotic yogurt for four weeks showed an improvement in emotional responses as measured by brain scans (2). A subsequent study by Mohammadi et al. (3) investigated the impacts of probiotic yogurt and probiotic capsules over 6 weeks and found a significant improvement in depression-anxiety-stress scores in subjects taking the specific strains of probiotics contained in the yogurt or capsules. Other studies with probiotics have indicated improvements in depression scores, anxiety, postpartum depression and mood rating in an elderly population (4-7).
Other studies have indicated a benefit of probiotic supplementation in alleviating symptoms of stress. In particular, researchers have looked at stress in students as they prepared for exams, while also evaluating other health indicators such as flu and cold symptoms (1). In healthy people, there is an indication that probiotic supplementation may help to maintain memory function under conditions of acute stress.
Introduction
Importance of Antioxidants in Consumer Products
Antioxidants are crucial ingredients in skincare products that combat oxidative stress and protect cells from damage caused by free radicals (1, 2, 3, 4). There are a few dominant pathways by which antioxidants remove free radicals, including hydrogen atom transfer (HAT), single electron transfer (SET), and sequential proton-loss electron transfer (SPLET) (5, 6).
HAT is one of the heavily investigated scavenging pathways for natural polyphenol antioxidants (5, 7). In this pathway, free radicals are neutralized by hydrogen atoms resulting from the cleavage of the O−H bond in phenolic hydroxyl groups. The energy associated with this bond cleavage, known as the bond dissociation energy (BDE), serves as a key indicator of antioxidant activity (8). Antioxidants with lower hydrogen BDE values often correspond to superior free radical scavengers due to their higher propensity to donate hydrogen atoms that neutralizes free radicals (4, 7).
BDEs can be measured experimentally through spectroscopic or calorimetric approaches by observing shifts in the absorption spectrum, bond vibrational frequencies, or measuring heat change associated with the bond breaking reaction (9). However, experimentally evaluating BDEs for the large design space of antioxidants is often cost-prohibitive, necessitating alternative computational approaches like quantum mechanics (QM) and machine learning (ML) to enable rapid screening of antioxidants.
Quantum Mechanics and Machine Learning Approach for High-throughput Screening
QM calculations offer a fast alternative compared to traditional experimental methods, which can measure BDEs by computing energies of a bonded pair of atoms relative to the energies of individual atoms in isolation. QM calculations have been used in studies to generate BDE datasets for characterizing antioxidant molecules, (8) where the QM calculated BDEs were found to have direct correlations with radical scavenging ability characterized by experiments (8,10). While BDEs can be determined using QM calculations, the computational cost of accurate QM calculations scales rapidly with the number of atoms, generally around O(N3) (11). This scaling relation limits their application in high-throughput screening of complex natural products with large number of atoms.
ML algorithms offer a possible solution to estimating BDEs of large molecules by training a model to predict QM-generated BDEs using molecular structure as input. Previous work focused on training ML models with hand-crafted features of the hydrogen sites, such as atomic distances, to classify which hydrogens were reactive or non-reactive (10). While informative, expert-defined features have shown promising model accuracy especially in the low data region, (12,13) the current state-of-the-art models are graph convolutional neural networks (GCNN) that represent a molecule as a graph, where atoms are nodes and bonds are edges. GCNNs are advantageous because they autonomously learn relevant features through convolution operations based on the atoms and connectivity of the molecule, which provides accurate structure-property relationships without having to pre-define descriptors (14). GCNNs have shown high accuracy in predicting properties of small drug-like molecules, (14,15) critical micelle concentrations of surfactants, (16, 17) viscosity of single or binary solvents, (12, 18, 19) and many others, (13) which demonstrates their broad applicability in computational chemistry. For predicting BDEs, previous work found that GCNNs can accurately predict BDE energies when trained with ~42 K small organic molecules (10, 20). While previous work have demonstrated that GCNNs can rapidly predict BDE energies, the transferability of these models applied to large antioxidant molecules is not well-explored in part due to the expense of the QM calculations cost to measure these BDEs. Inspired by prior work (20, 21) and our recent study demonstrating that atomic GCNNs can accurately predict acid dissociation constants (22) (pKa), we focus on training atomic GCNNs using an expanded dataset that includes large antioxidant molecules to create a model that can generalize BDE predictions for antioxidants. By combining QM and ML approaches, this work enables the high-throughput screening of antioxidants with promising BDEs.
To assess the effectiveness of antioxidants based on hydrogen BDEs, we combine QM and ML tools to train a model that can predict BDEs of antioxidants. First, we generate a database of antioxidant molecules and compute BDEs with QM calculations. Then, we train GCNNs to predict BDE with molecular structure as input using a large dataset with representative antioxidant molecules as part of the training set. We then demonstrate that GCNNs trained on small molecules do not extrapolate to larger antioxidant examples, and the inclusion of antioxidant molecules as part of the training set is important to create generalizable BDE models relevant for antioxidant screening.
Methods
Computing Bond Dissociation Energies with Density Functional Theory
The BDE of hydrogen atoms was calculated using the Jaguar quantum chemistry software within the Schrödinger Suite (23) (Version 2024-2). The "Bond and Ligand Dissociation" panel was used to compute BDEs using the functional theory and basis set of B3LYPand LACV3P** respectively (23, 24). The B3LYP is a hybrid functional that provides better accuracy compared against pure DFT methods. In comparison, the basis set LACV3P** has consistent performance across different elements compared with other choices like 6-31G**. For hybrid functional approaches, the compute costs scales approximately from O(N4) to O(N5) (25). While alternative higher levels of functional theory/basis sets could enhance BDE accuracy, their computational cost becomes prohibitive when applied to large molecules, rendering them impractical for generating an extensive dataset of BDEs (10, 25).
Bond Dissociation Energy Datasets
This study utilized two BDE datasets: a small molecule dataset and an antioxidant dataset. The small molecule dataset, obtained from the literature, comprises 41,379 neutral small molecules containing only C, H, N, & O with less than 10 heavy atoms (20). The antioxidant dataset consists of molecules of antioxidant molecules constructed from the COCONUT natural product database (26, 27). These molecules are restricted to the flavonoid group and have a maximum of 30 heavy atoms, resulting in a total of 2,693 molecules. Example molecules from the small molecule and antioxidant datasets are shown in Fig. 1A and 1B, respectively. Comparing Fig. 1A and Fig. 1B, antioxidants have more complex structures with several aromatic rings as compared to small molecules. Fig. 1C shows the molecular weight distribution between small molecule and antioxidant datasets, which highlights that the molecular weight for the antioxidant dataset is about double that of the small molecule dataset.
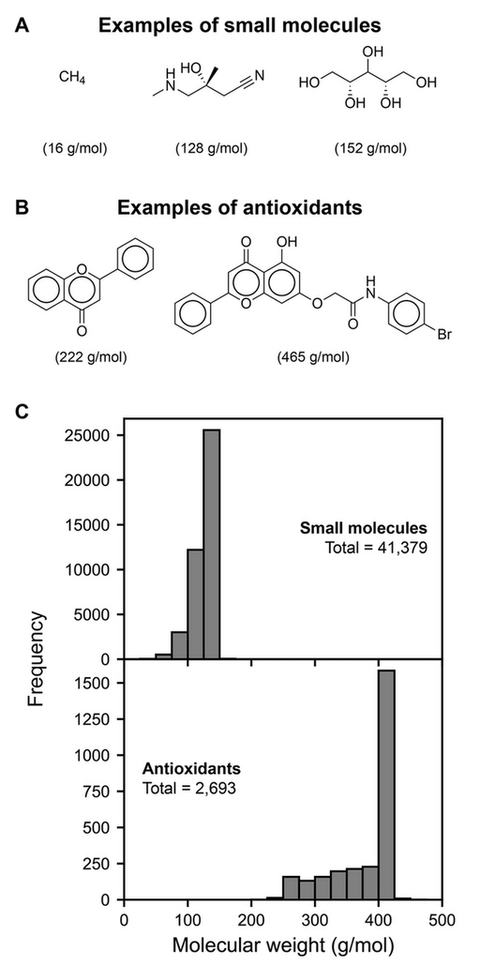
Figure 1. Examples of structures from (A) small molecule and (B) antioxidant dataset. Molecular weight in g/mol for each structure is shown in parenthesis. (C) Molecular weight distribution of small molecule and antioxidant datasets.
Machine Learning Models
QM-calculated molecular structures and BDE energies were used to train machine learning models for predicting BDE energy based on molecular structure, as shown in Fig 2. GCNNs were employed, representing molecules as graphs with atoms as nodes and bonds as edges (28). GCNNs have demonstrated success in materials science and chemistry, including BDE prediction and reaction free energy prediction (28, 29). Their advantage lies in autonomously generating relevant features through convolutional operations that aggregate information between neighboring atoms.
The GCNN architecture used in this study was adapted from a pKa prediction model that accurately predicted an acid's dissociation constant (22). In the GCNN architecture, each heavy atom is characterized by 74 atomic features, including one-hot-encoding of elements, valence or radical electron count, formal charge, hybridization, aromaticity, and explicit hydrogen count (22). Three graph convolution and update operations are performed, and the readout is fed into a dense neural network for BDE energy prediction. These operations allow the model to aggregate topological neighboring up to six bonds away from the bond dissociation site. The Pytorch version of GCNN (TorchGraphConv) was used as the model architecture (30).
Then the GCNNs were trained using the DeepAutoQSAR framework, Schrödinger's automated molecular property prediction engine (15, 31). This framework employs Bayesian optimization to select optimal model hyperparameters (e.g., number of neurons, epochs, dropout, etc.) based on model performance during a five-fold cross-validation procedure (5-CV). In 5-CV, each fold is partitioned into an 80:20 train:validation set, and the model is trained and evaluated on the validation set. This process is repeated until all examples have been part of the validation set exactly once. Performance is evaluated by measuring the coefficient of determination (R2) of all validation set examples. The final model comprises an ensemble of three GCNNs with the highest 5-CV R2 among all trained models. The average predictions of these GCNNs are reported, and prediction uncertainty is estimated by calculating the standard deviation of the predictions.
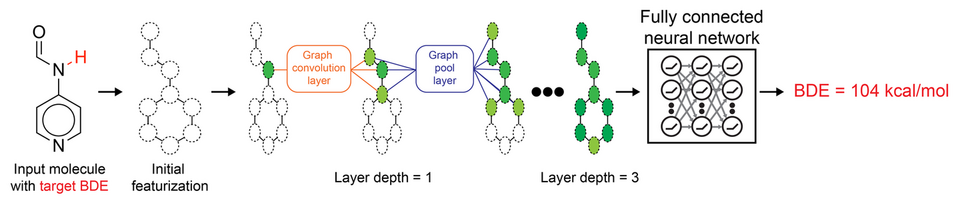
Figure 2. Machine learning workflow for predicting bond dissociation energies (BDE). The input molecule is represented as a graph with 74 atomic features. Three graph convolution and pooling operations are performed to aggregate topological information up to six bonds away from the bond dissociation site, which are then outputted to a readout layer and fully connected neural network to predict the BDE value.
Results
Distribution of Bond Dissociation Energies from Density Functional Theory
Fig. 3 presents the QM-calculated BDE distribution for both the small molecule and antioxidant datasets. This distribution reveals that the BDE values for both datasets are comparable, ranging from approximately 60 to 140 kcal/mol. For the small molecules dataset, the peak is centered around 90-100 kcal/mol. However, we observed two distinct peaks for the antioxidants dataset: the first peak at around 80-90 kcal/mol, and the second peak at around 100-110 kcal/mol. For antioxidants, the BDE values show a first peak close to 80 kcal/mol, which represent antioxidants with promising free radical neutralization activity given the low BDE values. The presence of multiple peaks in the antioxidant dataset highlights the diverse range of BDE values associated with different types of antioxidant molecules, which can be attributed to the complex structural characteristics of the antioxidant molecules.
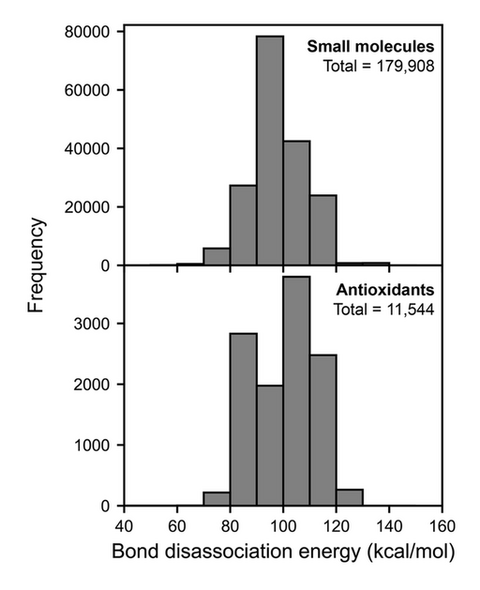
Figure 3. Bond dissociation energy (BDE) of small molecule and antioxidant datasets. The total number of BDEs is shown in the plot.
Machine Learning Predictions of Bond Dissociation Energies for Small Molecules
To evaluate the transferability of our baseline GCNN model, we initially trained it using only the small molecule dataset. This allowed us to assess whether the model could accurately predict the BDEs of the antioxidant dataset. To ensure a fair evaluation, we created separate test sets for each dataset. The small molecule test set comprised 4,349 BDE examples, while the antioxidant test set consisted of 2,286 BDE examples.
Fig. 4A presents the parity plot between the predicted and actual BDE values for predicting the small molecule test set using the baseline model. The results demonstrate that the BDE energies of small molecules were effectively captured, achieving a high test set R2 of 0.99 between predicted and actual values. This indicates that the baseline model can accurately predict the BDEs of small molecules. In contrast, Fig. 4B shows the parity plot between predicted and actual BDE values for predicting the antioxidant dataset using the same baseline model. Here, we observe a lower test set R2 of 0.81 compared to Fig. 3A. This suggests that the baseline model, trained solely on small molecules, cannot effectively generalize its predictions to large, structurally complex antioxidant molecules.
The discrepancy in performance between the small molecule and antioxidant datasets can be attributed to several factors. As highlighted in Fig. 1, there are significant differences in molecular weight and functional groups between the two datasets. These differences pose challenges for the baseline model, which was trained on a dataset with a narrower range of molecular properties. Consequently, the model struggles to extrapolate its predictions to the more diverse antioxidant dataset.
Machine Learning Predictions of Bond Dissociation Energies for Antioxidants
To enhance the model's accuracy, we retrained the GCNN by incorporating additional training data from the antioxidant dataset. Specifically, we augmented the small molecule dataset with 9,258 BDEs from the antioxidant dataset, representing approximately 80% of the antioxidant data. This expanded dataset allowed the GCNN to learn from a more diverse range of molecular structures and properties. We refer to this retrained model as the "antioxidant model."
Figure 4C presents the parity plot between predicted and actual BDE values when predicting the small molecule test set using the antioxidant model. The results indicate that the model maintains its high performance on small molecules, achieving a test set R2 of 0.99. This demonstrates that the inclusion of antioxidant data does not compromise the model's ability to accurately predict BDEs for small molecules. More importantly, Figure 4D shows the parity plot between predicted and actual BDE values when predicting the antioxidant test set using the antioxidant model. Here, we observe a significant improvement compared to the baseline model, with a test set R2 of 0.94. This result highlights the effectiveness of incorporating antioxidant data into the training process, enabling the model to generalize its predictions to large, complex antioxidant molecules.
The improved performance of the antioxidant model can be attributed to the inclusion of larger, more structurally complex molecules in the training data. As illustrated in Fig. 1, there is a substantial difference in molecular weight distribution between the small molecule and antioxidant datasets. Antioxidant molecules are typically two to three times larger than small molecules, and they often contain multiple rings and functional groups. These structural features can significantly influence the BDE of a molecule. By incorporating a diverse set of antioxidant molecules into the training data, the GCNN learns to recognize and account for these structural complexities, resulting in more accurate BDE predictions for antioxidants.
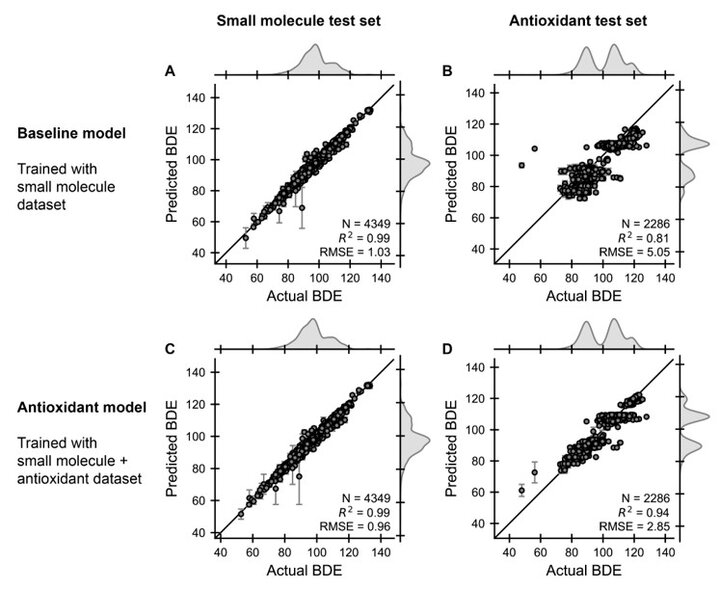
Figure 4. Parity plots between predicted and actual BDEs after training machine learning models with either the small molecule dataset (called baseline model) or the combination of small molecule and antioxidant dataset (called antioxidant model). Using the baseline model, the performance on a left-out test set is shown for (A) small molecule examples and (B) antioxidant examples. Similar test set performance plots are shown for the antioxidant model for (C) small molecule examples and (D) antioxidant examples. Statistics, such as the number of data points (N), coefficient of determination (R2) and root-mean-squared error (RMSE), are shown in the bottom right of each plot. All BDEs are reported in units of kcal/mol.
Discussion
Benefits of the QM-ML Approach
The results of this study highlight the strengths and potential of combining high-throughput QM calculations and machine learning techniques to identify antioxidants with promising properties (32). QM calculations possess the capability to rapidly compute a wide array of molecular properties that exhibit a strong correlation with experimental observations. To improve the model, the further study could incorporate other QM calculated properties into the datasets, like HOMO/LUMO, electron affinity, and ionization potentials, which correlate to other scavenging pathways. These QM-derived properties can help create large, comprehensive datasets that can be used to benchmark and refine machine learning workflows. By employing a consistent computational workflow to estimate BDEs, the risk of introducing errors arising from variations in experimental methods or conditions is mitigated. This work highlights that ML models can have poor generalizability or potentially overfit to specific chemistries based on the diversity of the training set, where a model trained with only small molecules may not translate to accurate predictions of large molecules (Fig. 4). The QM dataset can be used to rapidly expand the dataset to encompass a larger chemical space, and the ML model can be iteratively retrained to enhance its performance through a continuous feedback loop. By employing an iterative training approach, machine learning models would have a broader chemical coverage, which is a subject of future work. As demonstrated in this study, the strategic inclusion of antioxidant datasets significantly improves model performance compared to models trained exclusively on small molecule datasets. While these computational tools enable screening of antioxidants, downstream experimental validation is necessary to ensure the antioxidants have high activity.
Future Investigation Towards Formulation Optimization
While the current model could be used as the screening procedure to identify potential antioxidant candidates with high free radical scavenging capabilities, the top-performing candidates may not necessarily translate to be used in a final product due to the possible interactions between antioxidants and other ingredients that could impact product stability, shelf life, and so on. Instead of focusing on single-molecule properties, downstream formulation-property relationships that can optimize compositions of ingredients using machine learning models can be useful to screen antioxidant candidates and identify mixtures with promising properties. Formulation-property approaches have been shown to be broadly applied to various materials science domains, such as consumer products, pharmaceutical formulations, and petroleum applications (18). Future work will focus on passing antioxidant candidates from this work to downstream formulation studies with other common cosmetics ingredients like solvents, emulsifiers, and functional ingredients. These tools will enable the optimization of cosmetic formulations, which can lead to better consumer products with enhanced properties.
Conclusion
In this study, we have developed a robust computational workflow that leverages physics-based models and machine learning techniques to accurately predict the bond dissociation energy (BDE) of large antioxidant molecules commonly used in cosmetic applications. This workflow solely relies on the molecular SMILES pattern as input. The machine learning models achieve an impressive test set R2 of 0.94 for antioxidant molecules, significantly outperforming baseline models that yielded a lower test set R2 of 0.81 when trained exclusively on small molecules. These findings underscore the importance of incorporating large, structurally complex molecules into the training set to capture long-range interactions arising from molecular size and enhance the accuracy of BDE predictions.
Given an accurate antioxidant machine learning model, our workflow can be seamlessly extended to screen a vast library of antioxidants for desired BDE values. This capability holds immense promise for the cosmetic industry, as BDE serves as a key indicator of antioxidant functionality in cosmetic products. Moving forward, our research endeavors will focus on developing a generalizable BDE model that can effectively handle large molecules across diverse chemical spaces. Upon identifying the best antioxidant candidates from machine learning, the capability of validating the predicted BDE values against DFT calculations offers a way to robustly verify promising antioxidants before downstream experimentation. Additionally, we plan to employ feature-importance tools to gain deeper insights into how the atomic structure of antioxidants influences their BDE values. The screening of antioxidant structures can help down-select relevant ingredients that could be inputted into formulation-property machine learning modes, which can predict consumer good properties as a function of ingredients and compositions. Altogether, these computational tools pave the way for the data-driven design of antioxidant formulations with enhanced target properties.
Conclusion
The future of cosmetics lies in the continued evolution of holistic approaches which represents a transformative shift in the industry, merging scientific advancements, natural ingredients, and wellness principles. By understanding and embracing the interconnectedness of these elements, the cosmetics industry can cultivate products that not only enhance external beauty but also contribute to the overall well-being of individuals and the planet.
The interplay between beauty from within and topical cosmetics is the key for future products. The integration of biotechnology and green chemistry is revolutionizing cosmetic formulations, offering sustainable and biocompatible alternatives.
Developers can implement blockchain to trace the journey of ingredients from source to product. Nevertheless, the efficacy of the natural products should be scientifically proven. Marketers can communicate transparency as a brand value, and parallelly educate consumers by highlighting how specific ingredients contribute to radiant and healthy skin.
By embracing the synergy between these approaches and leveraging scientific advancements, the cosmetics industry can provide consumers with comprehensive beauty solutions that cater to both internal and external dimensions of beauty.
Surfactant Applications

The application area lends itself particularly well to the use of AI. Active today in this area is the US company Potion AI (6). The company provides AI-powered formulation tools for beauty and personal care R&D. Their offerings include Potion GPT, next generation ingredient and formula databases and AI document processing. Potion’s work could have a significant impact on the entire surfactant value chain, from raw material suppliers to end consumers. By using their GPT technology, they can help target work toward novel surfactant molecules that have optimal properties for specific applications. By using their ingredient and formula databases, they can access and analyze a vast amount of data on surfactant performance, safety, and sustainability. By using their AI document processing, they can extract and organize relevant information from patents, scientific papers, and regulatory documents. These capabilities could enable Potion AI's customers to design and optimize surfactant formulations that are more effective, eco-friendly, and cost-efficient. A particularly interesting application for this type of capability is deformulation.
Deformulation is the process of reverse engineering a product's formulation by identifying and quantifying its ingredients. Deformulation can be used for various purposes, such as quality control, competitive analysis, patent infringement, or product improvement. However, deformulation can be challenging, time-consuming, and costly, as it requires sophisticated analytical techniques, expert knowledge, and access to large databases of ingredients and formulas.
AI can potentially enhance and simplify the deformulation process by using data-driven methods to infer the composition and structure of a product from its properties and performance. For example, AI can use machine learning to learn the relationships between ingredients and their effects on the product's characteristics, such as color, texture, fragrance, stability, or efficacy. AI can also use natural language processing to extract and analyze information from various sources, such as labels, patents, literature, or online reviews, to identify the possible ingredients and their concentrations in a product.
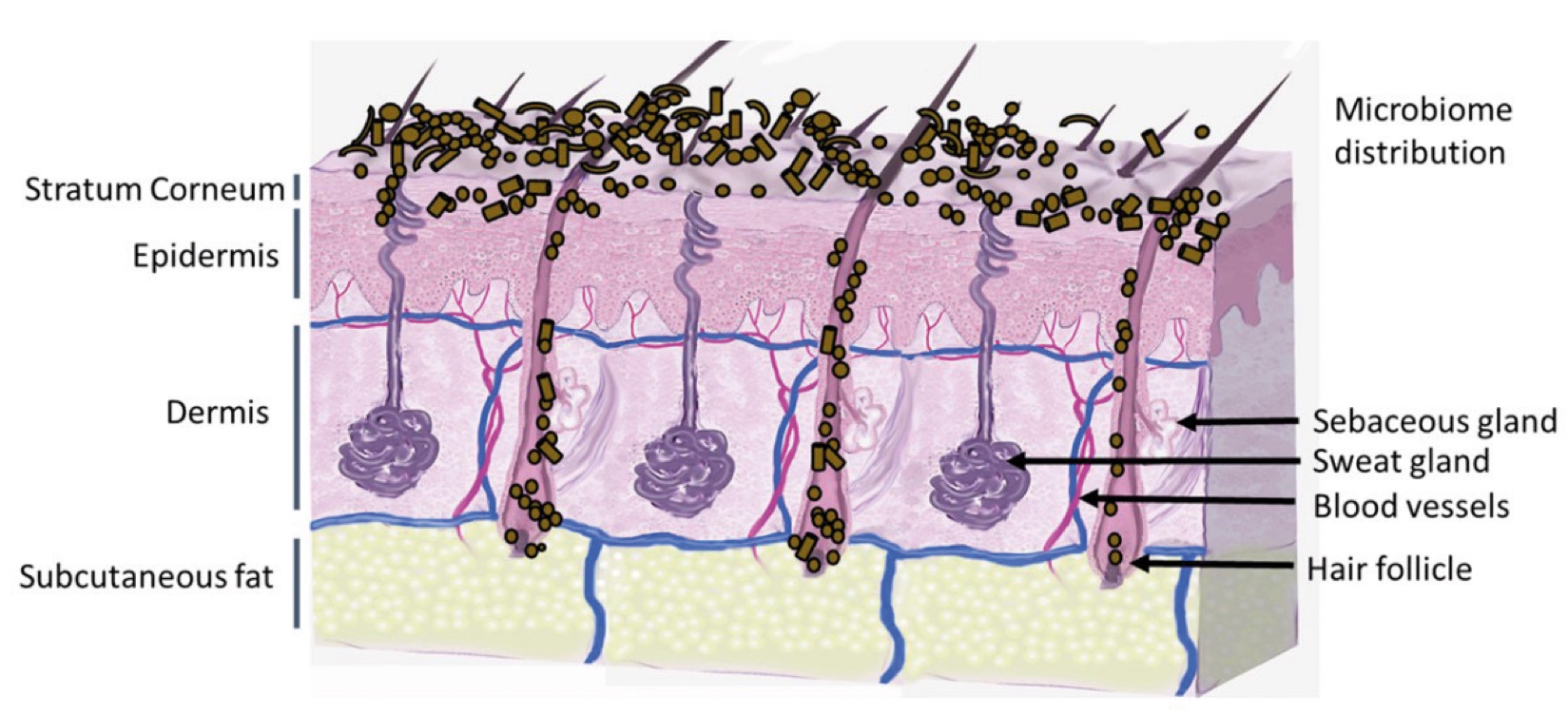
Figure 2. Skin Section with Microbiome. Most microorganisms live in the superficial layers of the stratum corneum and in the upper parts of the hair follicles. Some reside in the deeper areas of the hair follicles and are beyond the reach of ordinary disinfection procedures. There bacteria are a reservoir for recolonization after the surface bacteria are removed.
About the Author
Haidong Liu
Haidong Liu, PhD. CPG (Consumer Packaged Goods) Application Scientist from the Materials Science Group of Schrodinger since 2022. My expertise is in molecular modeling of complex systems. My work in Schrodinger consists of applying complex computational models to understand the science and help drive the innovations of personal care, cosmetics, and food products.
HAIDONG LIU
Schrödinger Inc, New York, NY, United States

References and notes
- Ak, M. Skin Care Creams: Formulation and Use. 2019.
- Vo, Q. V.; Nam, P. C.; Thong, N. M.; Trung, N. T.; Phan, C.-T. D.; Mechler, A. Antioxidant Motifs in Flavonoids: O–H versus C–H Bond Dissociation. ACS Omega 2019, 4 (5), 8935–8942. https://doi.org/10.1021/acsomega.9b00677.
- Galano, A.; Raúl Alvarez‐Idaboy, J. Computational Strategies for Predicting Free Radical Scavengers’ Protection against Oxidative Stress: Where Are We and What Might Follow? Int. J. Quantum Chem. 2019, 119 (2), e25665. https://doi.org/10.1002/qua.25665.
- Farrokhnia, M. Density Functional Theory Studies on the Antioxidant Mechanism and Electronic Properties of Some Bioactive Marine Meroterpenoids: Sargahydroquionic Acid and Sargachromanol. ACS Omega 2020, 5 (32), 20382–20390. https://doi.org/10.1021/acsomega.0c02354.
- Siddeeg, A.; AlKehayez, N. M.; Abu-Hiamed, H. A.; Al-Sanea, E. A.; AL-Farga, A. M. Mode of Action and Determination of Antioxidant Activity in the Dietary Sources: An Overview. Saudi J. Biol. Sci. 2021, 28 (3), 1633–1644. https://doi.org/10.1016/j.sjbs.2020.11.064.
- Galano, A.; Mazzone, G.; Alvarez-Diduk, R.; Marino, T.; Alvarez-Idaboy, J. R.; Russo, N. Food Antioxidants: Chemical Insights at the Molecular Level. Annu. Rev. Food Sci. Technol. 2016, 7 (1), 335–352. https://doi.org/10.1146/annurev-food-041715-033206.
- Mendes, R. A.; Almeida, S. K. C.; Soares, I. N.; Barboza, C. A.; Freitas, R. G.; Brown, A.; De Souza, G. L. C. Evaluation of the Antioxidant Potential of Myricetin 3-O-α-L-Rhamnopyranoside and Myricetin 4′-O-α-L-Rhamnopyranoside through a Computational Study. J. Mol. Model. 2019, 25 (4), 89. https://doi.org/10.1007/s00894-019-3959-x.
- Zhu, Q.; Zhang, X.-M.; Fry, A. J. Bond Dissociation Energies of Antioxidants. Polym. Degrad. Stab. 1997, 57 (1), 43–50. https://doi.org/10.1016/S0141-3910(96)00224-8.
- Blanksby, S. J.; Ellison, G. B. Bond Dissociation Energies of Organic Molecules. Acc. Chem. Res. 2003, 36 (4), 255–263. https://doi.org/10.1021/ar020230d.
- Muraro, C.; Polato, M.; Bortoli, M.; Aiolli, F.; Orian, L. Radical Scavenging Activity of Natural Antioxidants and Drugs: Development of a Combined Machine Learning and Quantum Chemistry Protocol. J. Chem. Phys. 2020, 153 (11), 114117. https://doi.org/10.1063/5.0013278.
- Schuch, N.; Verstraete, F. Computational Complexity of Interacting Electrons and Fundamental Limitations of Density Functional Theory. Nat. Phys. 2009, 5 (10), 732–735. https://doi.org/10.1038/nphys1370.
- Chew, A. K.; Sender, M.; Kaplan, Z.; Chandrasekaran, A.; Chief Elk, J.; Browning, A. R.; Kwak, H. S.; Halls, M. D.; Afzal, M. A. F. Advancing Material Property Prediction: Using Physics-Informed Machine Learning Models for Viscosity. J. Cheminformatics 2024, 16 (1), 31. https://doi.org/10.1186/s13321-024-00820-5.
- Jiang, D.; Wu, Z.; Hsieh, C.-Y.; Chen, G.; Liao, B.; Wang, Z.; Shen, C.; Cao, D.; Wu, J.; Hou, T. Could Graph Neural Networks Learn Better Molecular Representation for Drug Discovery? A Comparison Study of Descriptor-Based and Graph-Based Models. J. Cheminformatics 2021, 13 (1), 12. https://doi.org/10.1186/s13321-020-00479-8.
- Wu, Z.; Ramsundar, B.; Feinberg, E. N.; Gomes, J.; Geniesse, C.; Pappu, A. S.; Leswing, K.; Pande, V. MoleculeNet: A Benchmark for Molecular Machine Learning. Chem. Sci. 2018, 9 (2), 513–530. https://doi.org/10.1039/C7SC02664A.
- Benchmark Study of DeepAutoQSAR, ChemProp, and DeepPurpose on the ADMET Subset of the Therapeutic Data Commons, 2022. https://www.schrodinger.com/life-science/learn/white-papers/benchmark-study-deepautoqsar-chemprop-and-deeppurpose-admet-subset-therapeutic-data/.
- Qin, S.; Jin, T.; Van Lehn, R. C.; Zavala, V. M. Predicting Critical Micelle Concentrations for Surfactants Using Graph Convolutional Neural Networks. J. Phys. Chem. B 2021, 125 (37), 10610–10620. https://doi.org/10.1021/acs.jpcb.1c05264.
- Brozos, C.; Rittig, J. G.; Bhattacharya, S.; Akanny, E.; Kohlmann, C.; Mitsos, A. Predicting the Temperature Dependence of Surfactant CMCs Using Graph Neural Networks. J. Chem. Theory Comput. 2024, 20 (13), 5695–5707. https://doi.org/10.1021/acs.jctc.4c00314.
- Chew, A. K.; Afzal, M. A. F.; Kaplan, Z.; Collins, E. M.; Gattani, S.; Misra, M.; Chandrasekaran, A.; Leswing, K.; Halls, M. D. Leveraging High-Throughput Molecular Simulations and Machine Learning for the Design of Chemical Mixtures. January 8, 2025. https://doi.org/10.26434/chemrxiv-2024-4lff6-v4.
- Bilodeau, C.; Kazakov, A.; Mukhopadhyay, S.; Emerson, J.; Kalantar, T.; Muzny, C.; Jensen, K. Machine Learning for Predicting the Viscosity of Binary Liquid Mixtures. Chem. Eng. J. 2023, 464, 142454. https://doi.org/10.1016/j.cej.2023.142454.
- St. John, P. C.; Guan, Y.; Kim, Y.; Kim, S.; Paton, R. S. Prediction of Organic Homolytic Bond Dissociation Enthalpies at near Chemical Accuracy with Sub-Second Computational Cost. Nat. Commun. 2020, 11 (1), 2328. https://doi.org/10.1038/s41467-020-16201-z.
- Wen, M.; Blau, S. M.; Spotte-Smith, E. W. C.; Dwaraknath, S.; Persson, K. A. BonDNet: A Graph Neural Network for the Prediction of Bond Dissociation Energies for Charged Molecules. Chem. Sci. 2021, 12 (5), 1858–1868. https://doi.org/10.1039/D0SC05251E.
- Johnston, R. C.; Yao, K.; Kaplan, Z.; Chelliah, M.; Leswing, K.; Seekins, S.; Watts, S.; Calkins, D.; Chief Elk, J.; Jerome, S. V.; Repasky, M. P.; Shelley, J. C. Epik: P K a and Protonation State Prediction through Machine Learning. J. Chem. Theory Comput. 2023, 19 (8), 2380–2388. https://doi.org/10.1021/acs.jctc.3c00044.
- Bochevarov, A. D.; Harder, E.; Hughes, T. F.; Greenwood, J. R.; Braden, D. A.; Philipp, D. M.; Rinaldo, D.; Halls, M. D.; Zhang, J.; Friesner, R. A. Jaguar: A High‐performance Quantum Chemistry Software Program with Strengths in Life and Materials Sciences. Int. J. Quantum Chem. 2013, 113 (18), 2110–2142. https://doi.org/10.1002/qua.24481.
- Becke, A. D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98 (7), 5648–5652. https://doi.org/10.1063/1.464913.
- Bursch, M.; Mewes, J.; Hansen, A.; Grimme, S. Best‐Practice DFT Protocols for Basic Molecular Computational Chemistry**. Angew. Chem. 2022, 134 (42), e202205735. https://doi.org/10.1002/ange.202205735.
- Sorokina, M.; Merseburger, P.; Rajan, K.; Yirik, M. A.; Steinbeck, C. COCONUT Online: Collection of Open Natural Products Database. J. Cheminformatics 2021, 13 (1), 2. https://doi.org/10.1186/s13321-020-00478-9.
- Nainala, V. C.; Rajan, K.; Kanakam, S. R. S.; Sharma, N.; Weißenborn, V.; Schaub, J.; Steinbeck, C. COCONUT 2.0: A Comprehensive Overhaul and Curation of the Collection of Open Natural Products Database. September 10, 2024. https://doi.org/10.26434/chemrxiv-2024-fxq2s.
- Reiser, P.; Neubert, M.; Eberhard, A.; Torresi, L.; Zhou, C.; Shao, C.; Metni, H.; Van Hoesel, C.; Schopmans, H.; Sommer, T.; Friederich, P. Graph Neural Networks for Materials Science and Chemistry. Commun. Mater. 2022, 3 (1), 93. https://doi.org/10.1038/s43246-022-00315-6.
- Guha, R. D.; Vargas, S.; Spotte-Smith, E. W. C.; Epstein, A. R.; Venetos, M. C.; Wen, M.; Kingsbury, R. S.; Blau, S. M.; Persson, K. A. HEPOM: A Predictive Framework for Accelerated Hydrolysis Energy Predictions of Organic Molecules.
- Duvenaud, D.; Maclaurin, D.; Aguilera-Iparraguirre, J.; Gomez-Bombarelli, R.; Hirzel, T.; Aspuru-Guzik, A.; Adams, R. P. Convolutional Networks on Graphs for Learning Molecular Fingerprints.
- Yang, Y.; Yao, K.; Repasky, M. P.; Leswing, K.; Abel, R.; Shoichet, B. K.; Jerome, S. V. Efficient Exploration of Chemical Space with Docking and Deep Learning. J. Chem. Theory Comput. 2021, 17 (11), 7106–7119. https://doi.org/10.1021/acs.jctc.1c00810.
- Dinda, S.; Bhola, T.; Pant, S.; Chandrasekaran, A.; Chew, A. K.; Halls, M. D.; Sastry, M. Machine Learning-Based Design of Pincer Catalysts for Polymerization Reaction. J. Catal. 2024, 439, 115766. https://doi.org/10.1016/j.jcat.2024.115766.